



RT-qPCR được phát triển từ công nghệ PCR thông thường.Nó bổ sung các hóa chất huỳnh quang (thuốc nhuộm huỳnh quang hoặc đầu dò huỳnh quang) vào hệ thống phản ứng PCR truyền thống và phát hiện quá trình ủ và mở rộng PCR trong thời gian thực theo các cơ chế phát quang khác nhau của chúng.Sự thay đổi tín hiệu huỳnh quang trong môi trường được sử dụng để tính lượng sản phẩm thay đổi trong mỗi chu kỳ PCR.Hiện nay, các phương pháp phổ biến nhất là phương pháp nhuộm huỳnh quang và phương pháp thăm dò.
Phương pháp nhuộm huỳnh quang:
Một số thuốc nhuộm huỳnh quang, chẳng hạn như SYBR Green Ⅰ, PicoGreen, BEBO, v.v., không tự phát ra ánh sáng mà phát ra huỳnh quang sau khi liên kết với rãnh nhỏ của dsDNA.Do đó, khi bắt đầu phản ứng PCR, máy không phát hiện được tín hiệu huỳnh quang.Khi phản ứng chuyển sang giai đoạn ủ-mở rộng (phương pháp hai bước) hoặc giai đoạn mở rộng (phương pháp ba bước), lúc này các sợi kép được mở ra và DNA polymerase mới Trong quá trình tổng hợp sợi, các phân tử huỳnh quang được kết hợp trong rãnh nhỏ của dsDNA và phát ra huỳnh quang.Khi số chu kỳ PCR tăng lên, ngày càng có nhiều thuốc nhuộm kết hợp với dsDNA và tín hiệu huỳnh quang cũng liên tục được tăng cường.Lấy SYBR Green Ⅰ làm ví dụ.
Phương pháp thăm dò:
Đầu dò Taqman là đầu dò thủy phân được sử dụng phổ biến nhất.Có một nhóm huỳnh quang ở đầu 5′ của đầu dò, thường là FAM.Bản thân mẫu dò là một trình tự bổ sung cho gen mục tiêu.Có một nhóm dập tắt huỳnh quang ở đầu 3′ của chất huỳnh quang.Theo nguyên tắc truyền năng lượng cộng hưởng huỳnh quang (truyền năng lượng cộng hưởng Forster, FRET), khi nhóm huỳnh quang báo cáo (phân tử huỳnh quang cho) và nhóm huỳnh quang dập tắt (phân tử huỳnh quang nhận) Khi phổ kích thích trùng nhau và khoảng cách rất gần (7-10nm), sự kích thích của phân tử cho có thể gây ra sự phát huỳnh quang của phân tử nhận, trong khi quá trình tự phát huỳnh quang bị suy yếu.Do đó, khi bắt đầu phản ứng PCR, khi mẫu dò tự do và còn nguyên vẹn trong hệ thống, nhóm huỳnh quang báo cáo sẽ không phát ra huỳnh quang.Khi ủ, mồi và mẫu dò liên kết với mẫu.Trong giai đoạn mở rộng, polymerase liên tục tổng hợp chuỗi mới.DNA polymerase có hoạt tính exonuclease 5′-3′.Khi đến mẫu dò, DNA polymerase sẽ thủy phân mẫu dò khỏi mẫu, tách nhóm huỳnh quang báo cáo khỏi nhóm huỳnh quang dập tắt và giải phóng tín hiệu huỳnh quang.Vì có mối quan hệ một đối một giữa đầu dò và mẫu, nên phương pháp đầu dò vượt trội hơn phương pháp nhuộm về độ chính xác và độ nhạy của xét nghiệm.
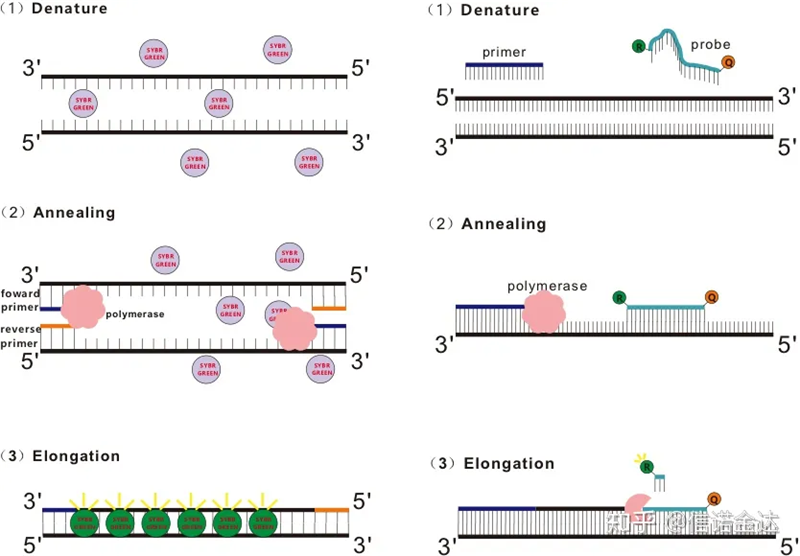
Hình 1 Nguyên tắc của qRT-PCR
thiết kế sơn lót
Nguyên tắc:
Các đoạn mồi nên được thiết kế trong vùng bảo tồn của chuỗi axit nucleic và có tính đặc hiệu.
Tốt nhất là sử dụng trình tự cDNA và trình tự mRNA cũng được chấp nhận.Nếu không, hãy tìm hiểu thiết kế vùng cds của chuỗi DNA.
Chiều dài của sản phẩm định lượng huỳnh quang là 80-150bp, dài nhất là 300bp, chiều dài mồi thường nằm trong khoảng 17-25 base và sự khác biệt giữa mồi ngược dòng và xuôi dòng không được quá lớn.
Hàm lượng G+C nằm trong khoảng từ 40% đến 60% và 45-55% là tốt nhất.
Giá trị TM nằm trong khoảng 58-62 độ.
Cố gắng tránh mồi dimer và self-dimer, cấu trúc kẹp tóc (không xuất hiện nhiều hơn 4 cặp bazơ bổ sung liên tiếp), nếu không thể tránh khỏi, hãy tạo ΔG<4,5kJ/mol* Nếu bạn không thể đảm bảo rằng gDNA đã bị loại bỏ trong quá trình sao chép ngược Làm sạch, tốt nhất là thiết kế các đoạn mồi có đầu intron *3′ không thể sửa đổi, và để tránh các vùng giàu AT, GC, tránh các đoạn mồi cấu trúc liên tục T/C, A/G (2-3) và không mồi
cụ thể Tương đồng của trình tự được khuếch đại không đồng nhất tốt hơn là ít hơn 70% hoặc có 8 tương đồng cơ sở bổ sung.
Cơ sở dữ liệu:
CottonFGD tìm kiếm theo từ khóa
Thiết kế mồi:
Thiết kế mồi IDT-qPCR
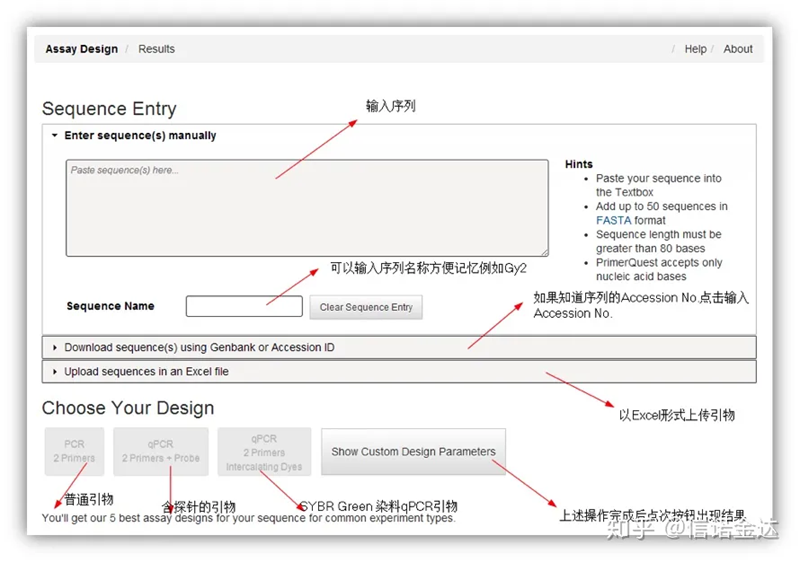
Fig2 Trang công cụ thiết kế mồi trực tuyến IDT
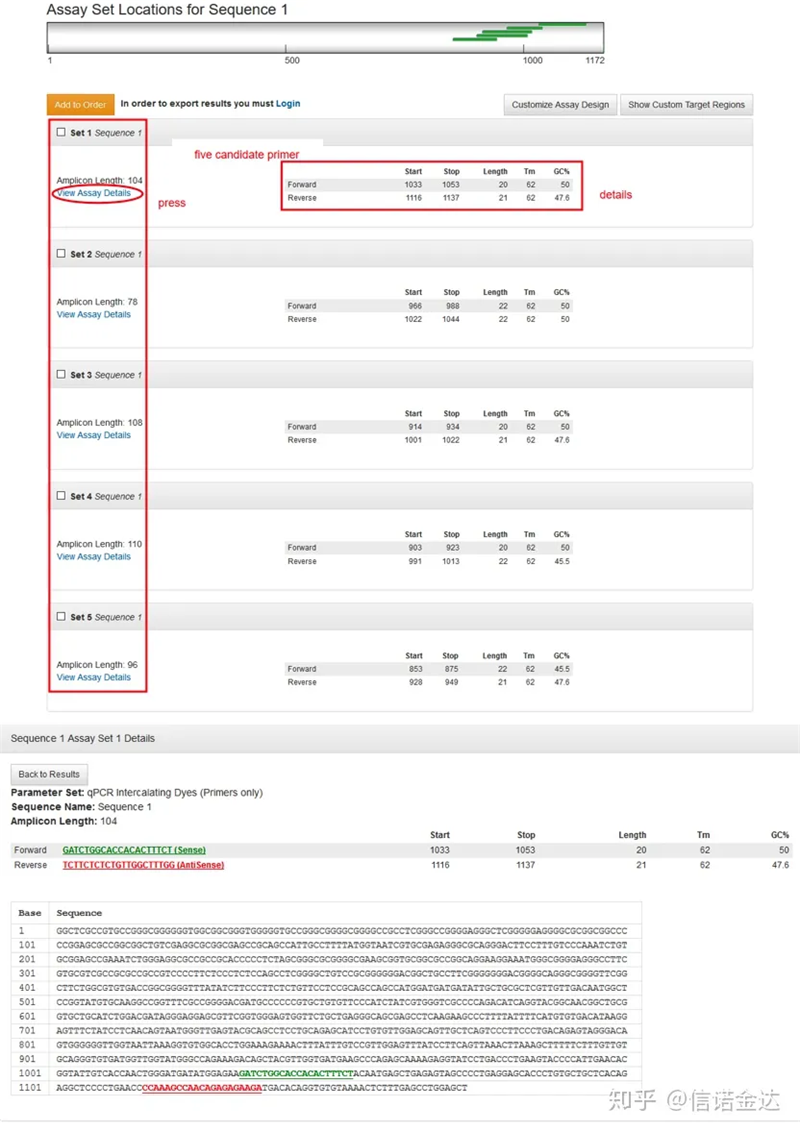
Hiển thị trang kết quả Fig3
Thiết kế mồi lncRNA:
lncARN:các bước tương tự như mRNA.
miARN:Nguyên tắc của phương pháp stem-loop: Vì tất cả các miRNA đều là các trình tự ngắn khoảng 23 nt nên không thể thực hiện PCR phát hiện trực tiếp, vì vậy công cụ trình tự stem-loop được sử dụng.Trình tự vòng lặp gốc là một DNA sợi đơn khoảng 50 nt, có thể tự tạo thành cấu trúc kẹp tóc.3' Phần cuối có thể được thiết kế như một trình tự bổ sung cho đoạn từng phần của miRNA, sau đó miRNA đích có thể được kết nối với trình tự vòng lặp gốc trong quá trình phiên mã ngược và tổng chiều dài có thể đạt tới 70bp, phù hợp với chiều dài của sản phẩm khuếch đại được xác định bởi qPCR.Thiết kế mồi miRNA đuôi.
Phát hiện cụ thể khuếch đại:
Cơ sở dữ liệu vụ nổ trực tuyến: Vụ nổ CottonFGD theo trình tự tương tự
Vụ nổ cục bộ: Tham khảo sử dụng Blast + để thực hiện vụ nổ cục bộ, linux và macos có thể trực tiếp thiết lập cơ sở dữ liệu cục bộ, hệ thống win10 cũng có thể được thực hiện sau khi cài đặt bash ubuntu.Tạo cơ sở dữ liệu vụ nổ cục bộ và vụ nổ cục bộ;mở bash ubuntu trên win10.
Chú ý: Bông nương và bông biển đảo là cây tứ bội nên bệnh đạo ôn kết quả thường từ 2 đọt trở lên.Trước đây, sử dụng NAU cds làm cơ sở dữ liệu để thực hiện blast có khả năng tìm thấy hai gen tương đồng chỉ với một vài điểm khác biệt về SNP.Thông thường, hai gen tương đồng không thể tách rời bằng thiết kế đoạn mồi, vì vậy chúng được coi là giống nhau.Nếu có indel rõ ràng, mồi thường được thiết kế trên indel, nhưng điều này có thể dẫn đến cấu trúc thứ cấp của mồi Năng lượng tự do trở nên cao hơn, dẫn đến giảm hiệu quả khuếch đại, nhưng điều này là không thể tránh khỏi.
Phát hiện cấu trúc thứ cấp mồi:
Các bước:mở oligo 7 → nhập trình tự mẫu → đóng cửa sổ phụ → lưu → xác định vị trí mồi trên mẫu, nhấn ctrl+D để đặt độ dài mồi → phân tích các cấu trúc thứ cấp khác nhau, chẳng hạn như phần thân tự thu nhỏ, dị vòng, kẹp tóc, không khớp, v.v. Hai hình ảnh cuối cùng trong Hình 4 là kết quả thử nghiệm của các mồi.Kết quả của mồi trước là tốt, không có cấu trúc mờ hơn và kẹp tóc rõ ràng, không có bazơ bổ sung liên tục và giá trị tuyệt đối của năng lượng tự do nhỏ hơn 4,5, trong khi mồi sau cho thấy liên tục 6 bazơ là bổ sung và năng lượng tự do là 8,8;Ngoài ra, một bộ điều chỉnh độ sáng nghiêm trọng hơn xuất hiện ở đầu 3′ và một bộ điều chỉnh độ sáng của 4 bazơ liên tiếp xuất hiện.Mặc dù năng lượng tự do không cao nhưng 3′ dimer Chl có thể ảnh hưởng nghiêm trọng đến độ đặc hiệu và hiệu suất khuếch đại.Ngoài ra, cần kiểm tra các kẹp tóc, dị vòng và không khớp.
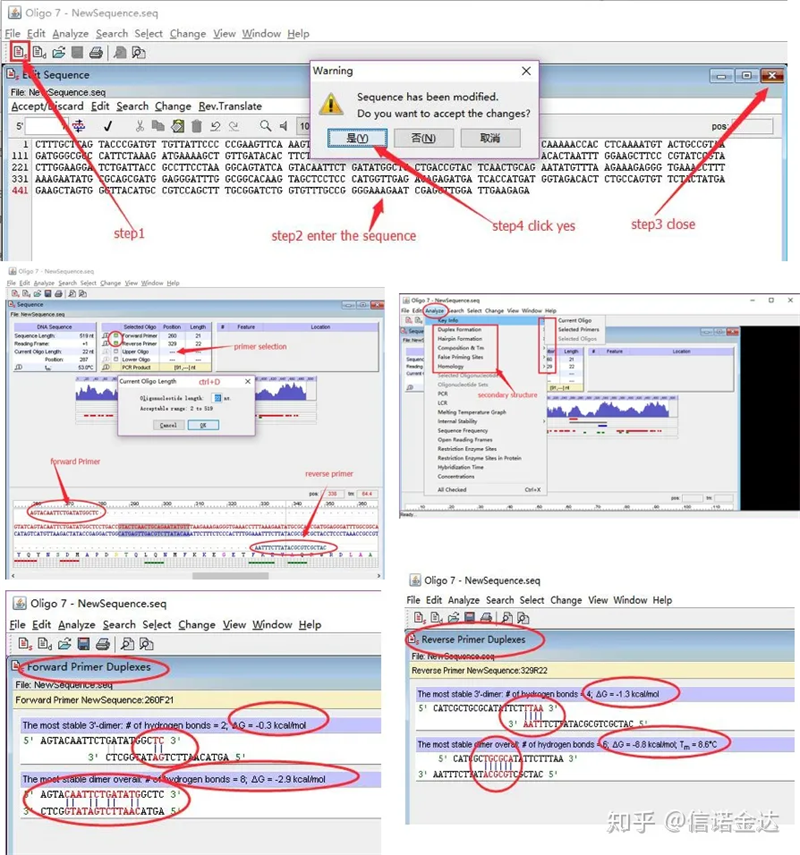
Kết quả phát hiện Fig3 oligo7
Phát hiện hiệu quả khuếch đại:
Hiệu quả khuếch đại của phản ứng PCR ảnh hưởng nghiêm trọng đến kết quả PCR.Cũng trong qRT-PCR, hiệu suất khuếch đại đặc biệt quan trọng đối với kết quả định lượng.Loại bỏ các chất, máy móc và quy trình khác trong bộ đệm phản ứng.Chất lượng mồi cũng có ảnh hưởng lớn đến hiệu quả khuếch đại của qRT-PCR.Để đảm bảo độ chính xác của kết quả, cả định lượng huỳnh quang tương đối và định lượng huỳnh quang tuyệt đối đều cần phát hiện hiệu quả khuếch đại của mồi.Người ta công nhận rằng Hiệu suất khuếch đại qRT-PCR hiệu quả là từ 85% đến 115%.Có hai phương pháp:
1. Phương pháp đường chuẩn:
Một.Trộn cDNA
b.Pha loãng độ dốc
c.qPCR
đ.Phương trình hồi quy tuyến tính để tính hiệu suất khuếch đại
2. LinRegPCR
LinRegPCR là chương trình phân tích Dữ liệu RT-PCR thời gian thực, còn được gọi là dữ liệu PCR định lượng (qPCR) dựa trên SYBR Green hoặc hóa chất tương tự.Chương trình sử dụng dữ liệu đã sửa không theo đường cơ sở, thực hiện hiệu chỉnh đường cơ sở trên từng mẫu riêng biệt, xác định cửa sổ tuyến tính và sau đó sử dụng phân tích hồi quy tuyến tính để điều chỉnh một đường thẳng thông qua bộ dữ liệu PCR.Từ độ dốc của đường này, tính hiệu quả PCR của từng mẫu riêng lẻ.Hiệu suất PCR trung bình trên mỗi bộ khuếch đại và giá trị Ct trên mỗi mẫu được sử dụng để tính toán nồng độ ban đầu trên mỗi mẫu, được biểu thị bằng đơn vị huỳnh quang tùy ý.Nhập và xuất dữ liệu thông qua bảng tính Excel.chỉ mẫu
trộn là cần thiết, không có gradient
các bước được yêu cầu:(Lấy Bole CFX96 làm ví dụ, không hoàn toàn Máy có ABI rõ ràng)
cuộc thí nghiệm:đó là một thử nghiệm qPCR tiêu chuẩn.
Đầu ra dữ liệu qPCR:LinRegPCR có thể nhận dạng hai dạng tệp đầu ra: RDML hoặc kết quả Khuếch đại định lượng.Trên thực tế, đó là giá trị phát hiện thời gian thực của số chu kỳ và tín hiệu huỳnh quang của máy và độ khuếch đại thu được bằng cách phân tích giá trị thay đổi huỳnh quang của hiệu suất phân đoạn tuyến tính.
lựa chọn dữ liệu: Về lý thuyết, giá trị RDML có thể sử dụng được.Người ta ước tính rằng vấn đề của máy tính của tôi là phần mềm không thể nhận ra RDML, vì vậy tôi có giá trị đầu ra excel là dữ liệu gốc.Trước tiên, nên thực hiện sàng lọc sơ bộ dữ liệu, chẳng hạn như lỗi thêm mẫu, v.v. Các điểm có thể bị xóa trong dữ liệu đầu ra (tất nhiên, bạn không thể xóa chúng, LinRegPCR sẽ bỏ qua các điểm này trong giai đoạn sau)
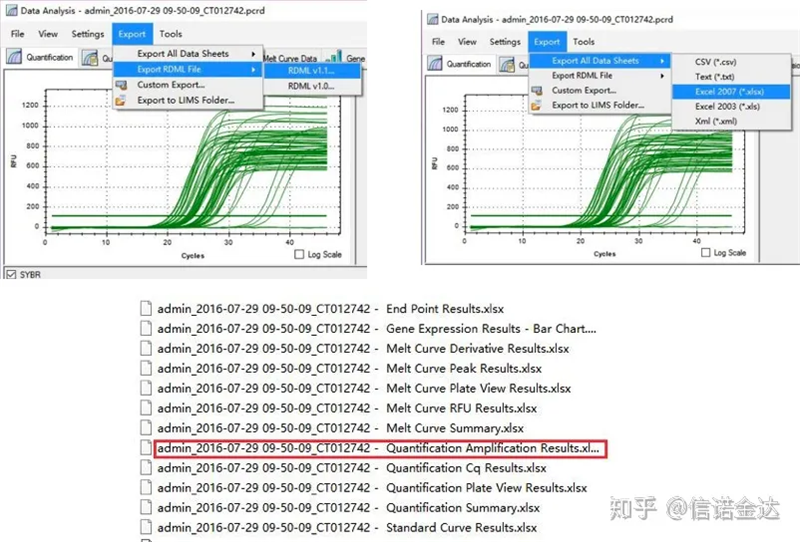
Fig5 xuất dữ liệu qPCR
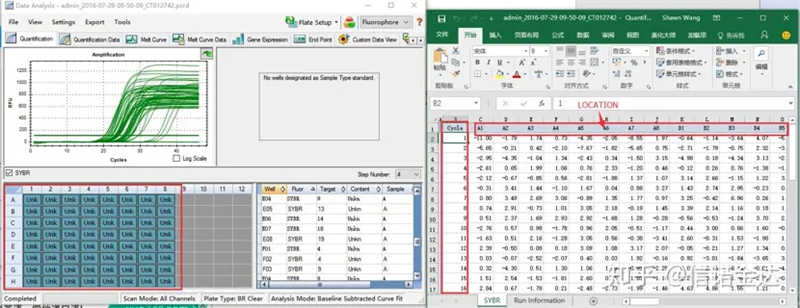
Fig6 lựa chọn các mẫu ứng cử viên
Dữ liệu đầu vào:Mở kết quả khuếch đại định tính results.xls, → mở tệp LinRegPCR → tệp → đọc từ excel → chọn tham số như trong Hình 7 → OK → nhấp vào xác định đường cơ sở
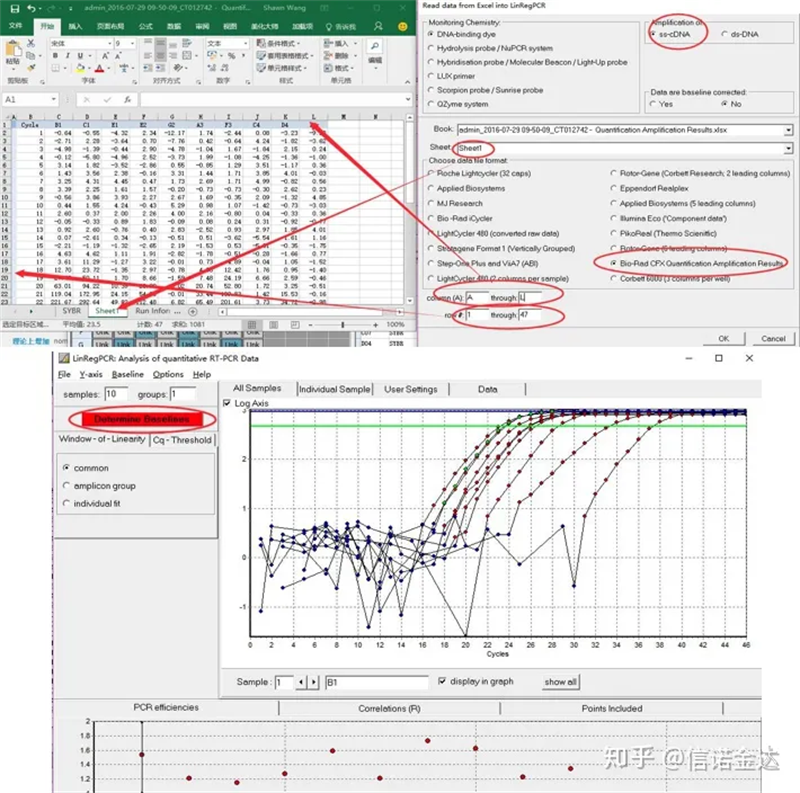
Fig7 các bước nhập dữ liệu linRegPCR
Kết quả:Nếu không có sự lặp lại thì không cần phân nhóm.Nếu có sự lặp lại, nhóm có thể được chỉnh sửa trong nhóm mẫu và tên của gen được nhập vào mã định danh, sau đó cùng một gen sẽ được nhóm tự động.Cuối cùng click vào file, xuất excel và xem kết quả.Hiệu suất khuếch đại và kết quả R2 của từng giếng sẽ được hiển thị.Thứ hai, nếu bạn chia thành các nhóm, hiệu suất khuếch đại trung bình đã hiệu chỉnh sẽ được hiển thị.Đảm bảo rằng hiệu suất khuếch đại của mỗi mồi nằm trong khoảng từ 85% đến 115%.Nếu nó quá lớn hoặc quá nhỏ, điều đó có nghĩa là hiệu quả khuếch đại của mồi kém.
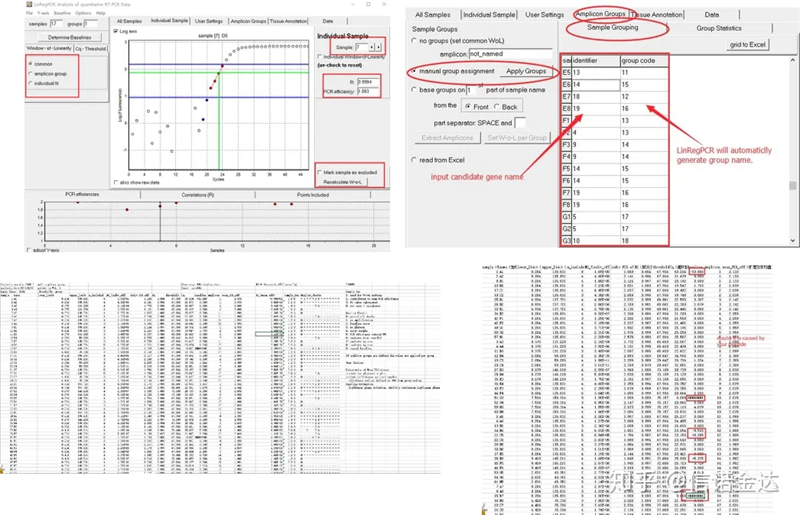
Hình 8 Kết quả và đầu ra dữ liệu
Quá trình thí nghiệm:
Yêu cầu chất lượng ARN:
độ tinh khiết:1.72.0 chỉ ra rằng có thể có isothiocyanate dư.Axit nucleic sạch A260/A230 phải ở khoảng 2 . Nếu có sự hấp thụ mạnh ở bước sóng 230nm, điều đó cho thấy có các hợp chất hữu cơ như ion phenate.Ngoài ra, nó có thể được phát hiện bằng điện di trên gel agarose 1,5%.Chỉ điểm đánh dấu, vì ssRNA không có sự biến tính và logarit trọng lượng phân tử không có mối quan hệ tuyến tính và trọng lượng phân tử không thể được biểu thị chính xác.Nồng độ: Về mặt lý thuyếtkhôngdưới 100ng/ul, nếu nồng độ quá thấp thì độ tinh khiết thường thấp không cao
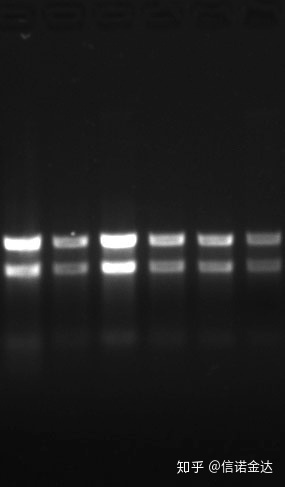
Gel RNA Fig9
Ngoài ra, nếu mẫu quý và nồng độ RNA cao, thì nên chiết tách mẫu sau khi chiết xuất và pha loãng RNA đến nồng độ cuối cùng là 100-300ng/ul để sao chép ngược.TRONGquá trình phiên mã ngược, khi mARN được phiên mã, đoạn mồi oligo (dt) có thể liên kết đặc hiệu với đuôi polyA được sử dụng để sao chép ngược, trong khi lncRNA và circRNA sử dụng đoạn mồi hexamer ngẫu nhiên (Random 6 mer) để sao chép ngược RNA tổng số. Đối với miRNA, đoạn mồi vòng cổ dành riêng cho miRNA được sử dụng để sao chép ngược.Nhiều công ty hiện đã tung ra bộ dụng cụ đuôi đặc biệt.Đối với phương pháp vòng lặp gốc, phương pháp đuôi thuận tiện hơn, thông lượng cao và tiết kiệm thuốc thử, nhưng Hiệu quả phân biệt các miRNA cùng họ không được tốt bằng phương pháp vòng lặp gốc.Mỗi bộ phiên mã ngược có các yêu cầu về nồng độ của các đoạn mồi đặc hiệu cho gen (stem-loops).Tham chiếu nội bộ được sử dụng cho miRNA là U6.Trong quá trình đảo ngược vòng lặp gốc, một ống U6 phải được đảo ngược riêng biệt và nên thêm trực tiếp các đoạn mồi trước và sau của U6.Cả CircRNA và lncRNA đều có thể sử dụng HKG làm tham chiếu nội bộ.TRONGphát hiện cDNA,
nếu không có vấn đề gì với RNA, thì cDNA cũng sẽ ổn.Tuy nhiên, nếu theo đuổi sự hoàn hảo của thí nghiệm, tốt nhất nên sử dụng gen tham chiếu bên trong (Gien tham chiếu, RG) có thể phân biệt gDNA với cds.Nói chung, RG là một gen giữ nhà., HKG) như Hình 10;Vào thời điểm đó, tôi đang tạo ra protein lưu trữ đậu tương và sử dụng intron chứa actin7 làm tài liệu tham khảo nội bộ.Kích thước của đoạn được khuếch đại của đoạn mồi này trong gDNA là 452bp, và nếu cDNA được sử dụng làm mẫu thì nó là 142bp.Sau đó kết quả kiểm tra phát hiện một phần cDNA thực sự đã bị gDNA làm ô nhiễm, đồng thời cũng chứng minh kết quả phiên mã ngược không có vấn đề gì, có thể dùng làm khuôn mẫu cho PCR.Việc chạy điện di trực tiếp trên gel agarose với cDNA là vô ích và nó là một dải khuếch tán, điều này không thuyết phục.
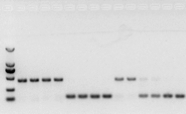
Hình 10 phát hiện cDNA
Việc xác định các điều kiện qPCRnói chung là không có vấn đề gì theo giao thức của bộ, chủ yếu ở bước giá trị tm.Nếu một số mồi không được thiết kế tốt trong quá trình thiết kế mồi, dẫn đến sự khác biệt lớn giữa giá trị tm và 60°C theo lý thuyết, thì cDNA sau khi trộn mẫu, nên chạy PCR gradient với mồi và cố gắng tránh đặt nhiệt độ không có dải làm giá trị TM.
Phân tích dữ liệu
Phương pháp xử lý PCR định lượng huỳnh quang tương đối thông thường về cơ bản là theo 2-ΔΔCT.Mẫu xử lý dữ liệu
Những sảm phẩm tương tự:
PCR thời gian thực dễ dàngTM –Taqman
PCR thời gian thực dễ dàngTM –SYBR XANH TÔI
RT Easy I (Master Premix để tổng hợp chuỗi cDNA đầu tiên)
RT Easy II(Master Premix để tổng hợp chuỗi cDNA đầu tiên cho qPCR)
Thời gian đăng: 14-03-2023



